

The first Endura Insights article laid out the motivation behind building Endura. In this second post, we go deeper into the chemistry at the heart of our approach.
The paradigm that built an industry doesn’t work for RNA
There are thousands of small molecule drugs on the market, nearly all targeting a protein. Most were developed from the same tried-and-true playbook: first purify your target and optimize drug molecules against it, then assess the best molecules in cells.
This approach is so ubiquitous that most people don’t appreciate that it relies on two fundamental assumptions. First, the target must have a defined region where a drug’s binding will disrupt function. Second, the target’s structure in a purified assay must reflect its structure inside a cell. Both assumptions have proven remarkably durable for proteins, due to the tight coupling of their structure and function. But for RNA, both assumptions break down.
Functionally, most proteins carry out their biology through specific active sites. A kinase has a pocket shaped to transfer a phosphate group, a protease has a cleft built to cut other proteins, and blocking those sites with a drug predictably disrupts function.[1] A messenger RNA has no equivalent. The functional region is the coding sequence, which the ribosome reads to produce protein, but small molecule binding there isn’t strong enough to stop the ribosome. For most mRNAs, there is no functional pocket where binding predictably interferes with biology.[2]
Structurally, proteins are molecular machines whose function depends on holding a precise 3D shape, and they have evolved remarkable stability to maintain it. That stability is what makes purified protein structures faithful to cellular reality, and what makes test-tube drug development work. mRNA is a message, not a machine. While each mRNA contains many structured domains, these are actively remodeled by helicases and reshaped by RNA-binding proteins that alter its accessible surface. The structure you observe in a test tube may not exist inside a cell, which means that a medicinal chemistry team can spend months optimizing against a binding site that isn't real.
The unique challenge of targeting RNA is that binding in a test tube does not predict function in the cell. Prior efforts to drug RNA with small molecules have made slow progress because the paradigm built for proteins fundamentally doesn’t apply.
Covalent chemistry offers a new way forward
We approached this challenge from a fresh perspective. What if the problem isn’t that RNA is undruggable, it’s that we’ve been using the wrong tools? Covalent chemistry offers a new way forward.
A covalent drug works in two steps. First, like any traditional small molecule, the drug recognizes a complementary surface on its target and binds reversibly. Then the reactive part of the molecule, called the covalent warhead, forms a permanent bond that locks the drug in place. This second step requires two conditions: the initial binding must hold the warhead in precisely the right geometry to react, and the warhead's chemistry must be compatible with the atom it reacts with.

Applied to RNA, covalent chemistry bridges the structure-function gap.
Reversible binding to the coding sequence can’t stop the ribosome, but a covalent bond can. It places a permanent chemical scar at a defined position on the mRNA, physically blocking the ribosome from reading through and shutting down production of the disease-causing protein. The drug doesn't need to find an active site, because the scar itself is the functional disruption.
Because the bond is permanent, you can screen compounds directly in living cells and read back exactly which sites were modified across every RNA.[3] Every hit identifies a real pocket, in the native RNA, that a chemist can optimize against.[4] A single experiment, run in the relevant cellular environment, reports target engagement and functional protein knockdown across the entire transcriptome.
This is what makes covalent chemistry a natural fit for RNA. To understand how far it can go, it helps to look at what it has historically unlocked for protein targets.
From serendipity to design
Covalent drugs aren't new. Aspirin, penicillin, and omeprazole all work by forming a permanent bond with their target. If you've ever taken aspirin for a headache, you've used a covalent drug.
For most of the twentieth century, the pharmaceutical industry deliberately avoided designing them. Medicinal chemistry teams routinely removed reactive compounds from their libraries and flagged them as liabilities. The worry was that a permanently reactive drug would inevitably hit the wrong targets and cause toxicity.
That changed when the first deliberately designed covalent drugs reached patients. Afatinib and Ibrutinib, both approved in 2013, were built from the start around the idea of forming a permanent bond with their targets, EGFR in lung cancer and BTK in B-cell malignancies. Ibrutinib became the first drug to successfully target BTK and has since treated nearly 300,000 patients worldwide. More recently, Paxlovid, the covalent antiviral prescribed to millions during the COVID-19 pandemic, was developed in months using the same design logic.[5]
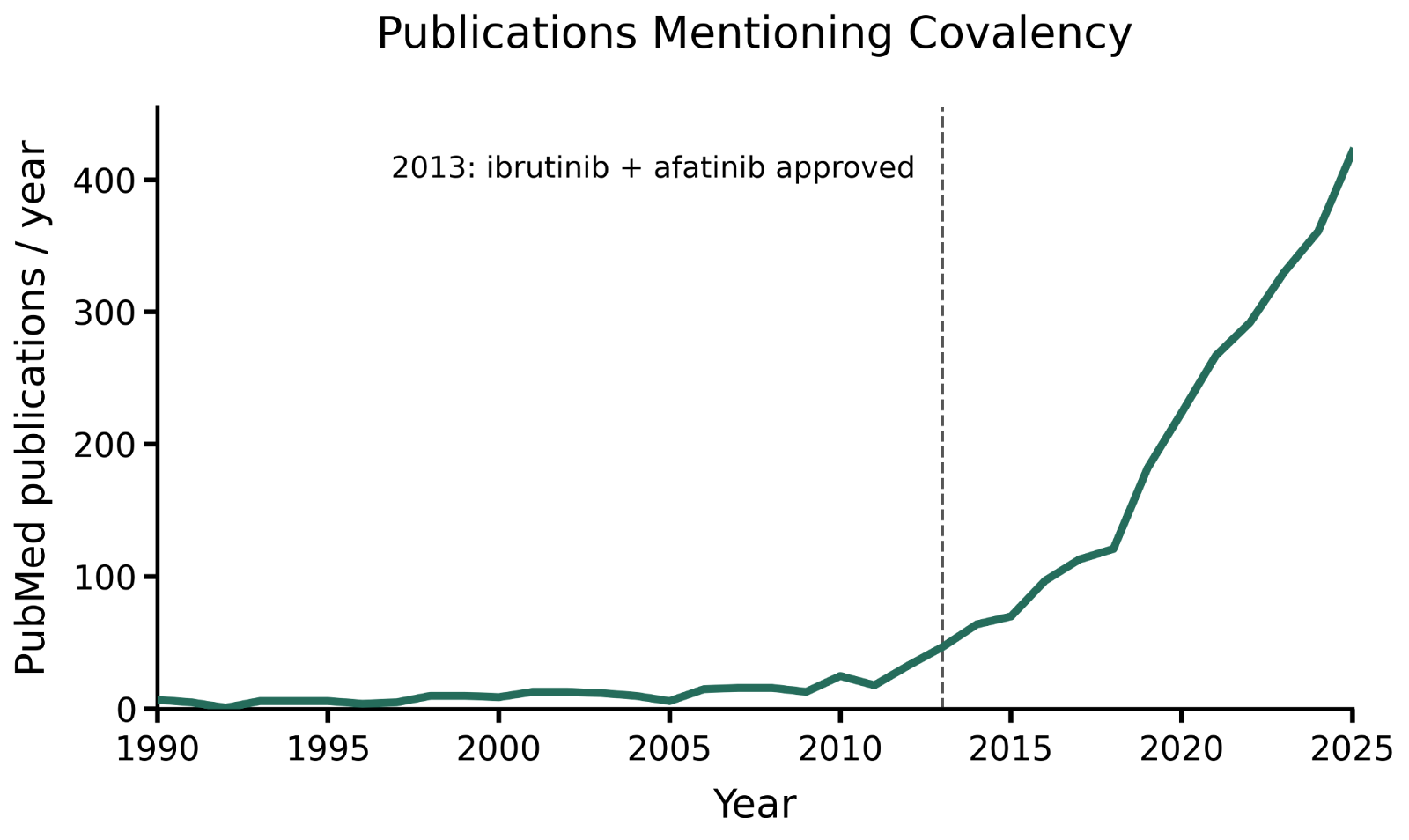
Today, over 50 FDA-approved drugs work through covalent mechanisms across oncology, immunology, and infectious disease.[6] Covalent drugs are now being developed for chronic conditions and brain indications, areas once considered off-limits for covalent chemistry.
Covalency solved a 40-year challenge
KRAS is the clearest example of what covalent chemistry can do when the traditional toolkit falls short. In 1982, KRAS was the first human oncogene ever discovered. Within a few years, it became clear that mutations in KRAS drove roughly a quarter of all cancers. Patients with KRAS-driven disease faced some of the worst outcomes in oncology, and a generation of drug hunters set out to stop it.
For nearly four decades, none of them succeeded.[7] The challenge with KRAS was its unusually smooth surface, which offers no obvious pocket for a small molecule to settle into. The one exception is its active site, but that pocket is occupied by its natural substrate, GTP, which binds so tightly that no reversible drug can displace it.
The breakthrough came in 2013 when Kevan Shokat's group at UCSF published a different approach. Instead of trying to block GTP, they focused on one specific mutant, KRAS G12C, which replaces a glycine with cysteine, the only amino acid covalent chemistry could reliably engage at the time.[8] They screened a library of 480 reactive compounds against the protein and discovered a cryptic pocket that only opened up once a compound had engaged the cysteine. That pocket sits away from the active site, and binding there locks KRAS in its inactive form without the drug having to compete with GTP.
Less than a decade later, two covalent KRAS G12C inhibitors, Sotorasib and Adagrasib, were FDA-approved for non-small-cell lung cancer and colorectal cancer. Both bind the pocket that Shokat's group discovered, with more drugs targeting the same site currently in clinical development.
The success of G12C inspired drug hunters to pursue other KRAS mutants, particularly G12D, the most prevalent mutation across cancer types. But G12D introduces an aspartate instead of cysteine, a far less reactive amino acid. In parallel, chemists had spent years building out a broader toolbox of covalent warheads (nitriles, aldehydes, sulfonyl fluorides, activated esters, and others) capable of engaging amino acids beyond cysteine. Revolution Medicines directly drew on that work. In January 2026, their G12D inhibitor Zoldonrasib received Breakthrough Therapy designation from the FDA.
The covalent toolbox is ready for RNA
The warheads that cracked G12D were part of a broader expansion to engage new amino acids, like lysine, tyrosine, and histidine. These amino acids carry reactive groups chemically similar to the ones on RNA, which means a warhead built to engage a lysine can often engage a comparable group on RNA.
This expanding covalent toolbox has, as a byproduct, produced the first generation of warheads capable of engaging RNA. These warheads are just a few years old, and few have connected them to RNA. The opportunity is enormous: hundreds of RNA targets could now be reachable by covalent drugs, providing treatments for diseases where current medicines fall short.
Endura is built on the bet that covalent chemistry makes RNA druggable. The permanent, enduring nature of the covalent bond is what inspired our name.